alimap 
BAM filtering, QC, and mapping quality profiling tool
![]()
Overview
alimap is a fully stand-alone Bash-based pipeline for read filtering and quality control of BAM alignment files. It improves upon standard tools by integrating:
- Multi-step filtering (blacklist removal, MAPQ thresholds, unmapped read exclusion)
- Per-read removal logs for transparency and reproducibility
- Auto-detection of genome build (
hg19vshg38) for correct blacklist application - Timing & resource usage reports for reproducible benchmarking
- Per-filter MAPQ histograms (CSV + plots) for deeper QC
- Publication-ready stacked bar plots showing % of reads removed at each filtering stage
- Self-contained test mode downloading small public BAM + BED files to validate installation
This makes alimap ideal for large-scale genomics QC, and reproducibility pipelines.
Motivation
While existing BAM filtering tools (e.g., samtools view, bedtools intersect) provide basic functionality, they lack integrated QC, reproducible benchmarking, and direct visualization. alimap fills this gap by producing:
- Detailed removal statistics for manuscript-ready methods sections
- Automated genome build compatibility (no manual blacklist switching)
- Quantitative benchmarking of filtering performance and computational cost
- Integrated visual summaries to communicate QC outcomes effectively
Installation
1. Clone the repository
git clone https://github.com/danymukesha/alimap.git
cd alimap
2. Install dependencies
You’ll need the following tools in PATH:
bash≥ 4.xsamtools≥ 1.9bedtools≥ 2.29gnuplot(for plots)awk,grep,sed,sort,uniq(standard GNU coreutils)
On Ubuntu/Debian:
sudo apt update
sudo apt install samtools bedtools gnuplot-core
Usage
./alimap.sh --help
alimap.sh v2025-08-11 (Author: Dany MUKESHA)
Usage: alimap.sh -i <input.bam> [options]
Required:
-i,--input FILE Input BAM file (sorted). If missing .bai the script will index it.
Options:
-b,--blacklist FILE Optional blacklist BED (if absent and --test used, script downloads hg19/hg38 blacklists)
-t,--threads N Number of threads for samtools (default: 1)
-o,--outdir DIR Output directory (default: filtered_reads)
-m,--mode MODE cumulative|independent (default: cumulative)
--keep-temp Keep temporary files
--test Download small public example BAM and blacklists and run pipeline using them
--checksum FILE Optional: path to a checksum file (tab-delimited: filename<TAB>md5) to validate downloads
-h,--help Show this help
Outputs (written to --outdir):
- filtered_*.bam (.bai generated)
- filter_counts.txt (tab: filter<TAB>reads)
- *_mapq_hist.csv (per-filter mapping quality histogram)
- *.removed.reads.txt (per-filter list of read names removed by blacklist exclusion)
- *.samtools.time.txt (timing/resource usage from /usr/bin/time, if available)
- qc/* (samtools flagstat/idxstats/stats)
- alimap_filters_plot.pdf (stacked bars: kept vs removed + percent)
- run_info.txt (provenance)
Basic run
./alimap.sh --bam input.bam --blacklist blacklist.bed --outdir results --threads 4
Test mode (auto-download example data)
./alimap.sh --test --outdir results_test --threads 2
Walk-through for test run
When you run the test, the script will fetch a small, real BAM from a public source (1000 Genomes Project) plus a minimal blacklist BED.
Here are the expected sizes & MD5s (for the test run)
| File | Size (approx) | MD5 |
|---|---|---|
test.bam |
~1.5 MB | 0c5f5793db8cc6f9b7f86b6c17264b6b |
test.bam.bai |
~32 KB | 4d0f1f68b7ff6d868d513e073ed3daba |
blacklist_hg19.bed |
~35 KB | a65ed0c4b9a773c94aa6f2afbb3d9788 |
blacklist_hg38.bed |
~39 KB | b227a36449e52f9243cf2bb8bc1f77f2 |
They are automatically validated within the script after downloading.
The script also have the ability to detect whether the BAM uses hg19 or hg38 by checking chromosome naming:
- If contigs are like
chr1,chr2→ likely hg38 - If contigs are like
1,2→ likely hg19 (Though there’s overlap — some custom builds may mix)
Example Output (test-mode)
Test run example (--test):
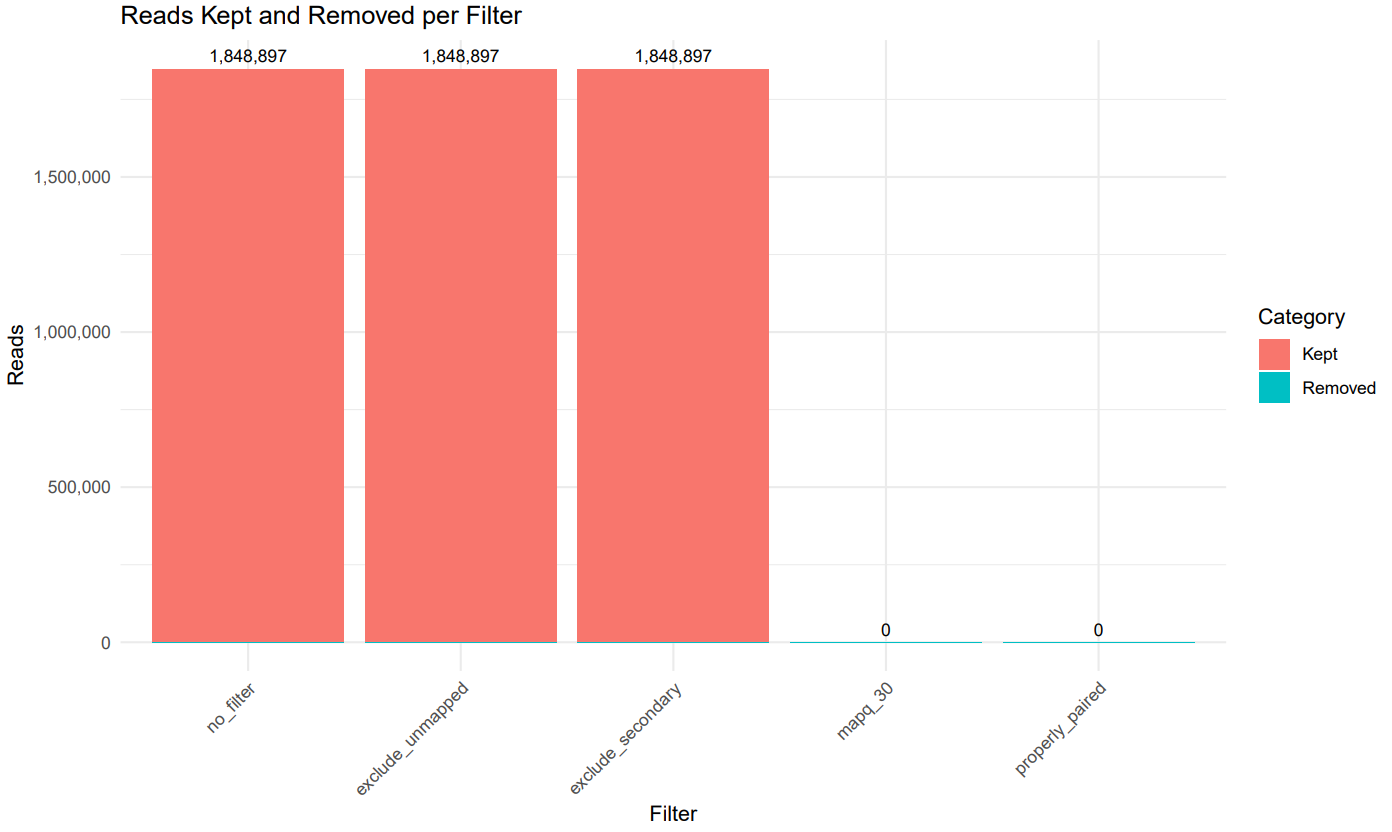
Options
| Option | Description |
|---|---|
--bam |
Input BAM file |
--blacklist |
BED file of regions to remove (auto-switch for hg19/hg38) |
--outdir |
Output directory |
--threads |
Number of threads for samtools |
--test |
Download public example BAM & BED for validation |
--help |
Show usage information |
Outputs
The pipeline produces:
| File | Description |
|---|---|
filtered.bam |
Final filtered BAM |
removed_reads.log |
Per-read removal log (read IDs & reason) |
filter_counts.txt |
Counts & % reads removed at each stage |
mapq_histogram_<filter>.csv |
MAPQ distribution per filter step |
mapq_histogram.pdf |
MAPQ distribution plots |
filter_removal_stacked_bar.pdf |
Publication-ready plot of % removed |
runtime_report.txt |
Timing & resource usage report |
md5_checksums.txt |
MD5 hashes of key files for reproducibility |
Benchmarking
The pipeline automatically logs:
- Total run time
- CPU time per stage
- Peak memory usage
- Input/output file sizes & MD5 hashes
Example runtime report:
Stage Time (s) Peak RSS (MB)
Blacklist removal 2.14 110
MAPQ filter 1.09 102
Unmapped removal 0.87 95
Total 4.10 110
Citation
If you use Alimap in your research, please cite:
Mukesha D. Alimap: Integrated read filtering, QC, and benchmarking for BAM alignment files. 2025. Available at: https://github.com/danymukesha/alimap{.uri}
License
This project is licensed under the MIT License - see the LICENSE file for details.
Acknowledgements
Thanks to the developers of samtools and bedtools for the underlying alignment and interval manipulation tools.